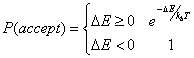Monte Carlo simulation (MCS) is a common methodology to
compute pathways and thermodynamic properties of proteins. A simulation run is a
series of random steps in conformation space, each perturbing some degrees of
freedom of the molecule. A step is accepted with a probability that depends on
the change in value of an energy function.
Typical energy functions sum many terms. The most costly ones
to compute are contributed by atom pairs closer than some cutoff distance. This
software uses a new method that speeds up MCS by efficiently computing the
energy at each step. The method exploits the facts that proteins are long
kinematic chains and that few degrees of freedom are changed at each step. A
novel data structure, called the ChainTree, captures both the kinematics and the
shape of a protein at successive levels of detail. It is used to find all atom
pairs contributing to the energy. It also makes it possible to identify partial
energy sums left unchanged by a perturbation, thus allowing the energy value to
be incrementally updated.
The method used in this software is based on
an algorithm for efficiently computing self-collisions in a deformable kinematic
chain [1]. This algorithm was extended and adapted for the
efficient computation of the internal energy of a protein as it undergoes
deformations during an MCS [2].
MCS of proteins can be performed in many
different ways. The methodology that is used is customized to match the goals of
the specific simulation at hand. Some of the aspects that are often
changed are:
Move set: The changes applied to
the protein structure at each step of the simulation.
Acceptance criterion: The rule
according to which new conformations are accepted or rejected.
Energy Function: The score that
is given to each conformation. This is often the internal energy of the
conformation, but it is not required to have a physical meaning.
While the software we provide implements only
one such MCS methodology, the underlying data structures can be used efficiently
with a large variety of move sets, acceptance criteria and energy functions. The
only restrictions are:
Only backbone and side-chain dihedral angles
can be changed, leaving the bond lengths and angles fixed throughout the
simulation.
The efficiency of the software drops as the
number of simultaneous backbone angles that are changed at each simulation step
is increased.
The energy computation is most efficient when
energy terms depend on the distances between pairs of atoms and a cutoff
distance is used to limit the number of pairs that contribute to the total
energy. Single parameter terms such as a dihedral angle potential can also be
used efficiently.
Our implementation can be used to run an MCS
using the choices we have made, which are described below. It can also be used
as an example for anyone that wants to use his/her own move set / acceptance
criterion. The energy function that is used can also be replaced, although that
may turn out to be more difficult.
The Software
This software implements the following MCS
methodology:
Move Set: There are two kinds of moves:
Backbone move: Three backbone angles are
changed simultaneously. Each angle is changed by a random amount drawn from a
zero-mean Gaussian distribution.
Rotamer move: A number of side-chain rotamers
are changed simultaneously to different rotamer values drawn from a fixed
library of rotamers (The backbone independent rotamer library of Dunbrack and
Cohen [3] from August '99).
Each step of the simulation is comprised of
one attempted backbone move followed by a number of attempted rotamer moves.
Acceptance criterion: The Metropolis
criterion:
Energy function: EEF1 of Lazaridis and
Karplus [4].
The different parameters of the simulation are
determined by the following command line options:
-I file-name
The input protein structure used
as the initial conformation of the simulation. The structure is described by
the backbone dihedral angles and the index of the rotamer of each amino
acid. For example:
ARG 100 140 5
TYR -60 70 3
ASN 44 -134 1
The first entry is the three letter amino acid code, then
come the Φ and Ψ backbone angles and finally
the index of the rotamer to be used. The file must have a ".angs" extension.
-T
temperature
The temperature of the
simulation. (Default is 298.15 K)
-S
step-number
The number of steps in the
simulation (Default is 100,000)
-N
save-interval
The number of steps between save
operations. Each save operation the current conformation is saved in a ".angs"
file as well as in a PDB style ".pdb" file. Also, the energy is saved in the
".out" file. (Default is 1000)
-K
backbone-angs
The number of backbone angles
that are changed simultaneously during a backbone move. (Default is 1)
-Aangle-stdev
The standard deviation (in
degrees) of the zero-mean Gaussian distribution from which the change to
each backbone angle is drawn. (Default is 10°)
-F
rotamer-moves
The number of rotamer moves
attempted during each simulation step. (Default is 5)
-R
changed-rotamers
The number of rotamers that are
changed each rotamer move. (Default is 1)
-O
output-files
The name of the output files
without any extension. The different extensions are added by the program. At
each save interval the current conformation and its energy value are saved.
The default is to save only intermediate energy values and the final
conformations.
-U
random-seed
The random seed that is used by
the programs random number generator. (Default is 0)
-C
reference-structure
A protein structure specified in
PDB style file with a ".pdb" extension that is used as the reference
structure to which all saved structures are compared using the cRMSD
similarity measure. The similarity measure of the current conformation is
saved together with its energy value. The default is to use the initial
conformation as the reference structure.
-H
home-dir
The directory where the
executable together with the files "rotamers.h", "rotamer_coords"
and "exclusions" reside.
-X
output-dir
The directory where intermediate
results will be saved.
Download
The software
source-code can be downloaded from here.
To build the executable
type make -k mcs
in the directory where you installed the source files.
References
Lotan, I.,
Schwarzer, F., Halperin, D., Latombe, J.C.: Efficient maintenance and
self-collision testing for kinematic chains. In: Symposium on Computational
Geometry (2002) 43–52. [PS],
[PDF]
Lotan, I.,
Schwarzer, F., Latombe, J.C.: Efficient Energy Computation for
Monte-Carlo Simulation of Proteins. In: Workshop on Algorithms in
Bioinformatics (2003), To appear. [PS],
[PDF]
Dunbrack Jr.,
R. L. and Cohen, F. E. Bayesian Statistical Analysis of Protein
Sidechain Rotamer Preferences ." Protein Science 6 (1997) 1661-1681.
Lazaridis, T.,
Karplus, M.: Effective energy function for proteins in solution. Proteins
35 (1999) 133–152.